|
by
Matthew Belmonte and Ruth Carper
in: Neuroimaging in Child Neuropsychiatric Disorders, edited by Bernard Garreau
Springer-Verlag, April 1998
(pages 157-171)
CITED IN:
Of all the developmental brain disorders, autism is perhaps the most fascinating, the most mysterious, and the most telling--fascinating, because of its impact on absolutely every aspect of a child's perception of and interaction with the surrounding world; mysterious, because of the complexity of the many interacting brain systems that it perturbs; telling, because it strikes at the social, cognitive, and linguistic abilities that seem, at least on the surface, so essential to one's very humanity. During the past few decades, the riddle of autism has begun to yield to advances in the study of autistic behavior and the biological foundations that affect this behavior. The examination of brain anatomy, physiology, histology and function in people with autism continues to supply new information on the nature and ætiology of this complex syndrome. This chapter will focus primarily on the neuroanatomical and neurophysiological abnormalities found in autistic subjects, particularly those seen in magnetic resonance imaging (MRI) and event-related potential (ERP) studies. Additional chapters will discuss evidence from other quantitative EEG measurements, magnetic resonance spectroscopy, and functional imaging (functional MRI and PET).
A PLETHORA OF PROPOSALS
Many regions of the cerebrum have been posited as loci of autism. In fact, autism impacts so many abilities and behaviors that it's difficult to find any brain region that has not been put forth as a possible site of dysfunction. Proposed sites have included the limbic system, brainstem, basal ganglia, vestibular system, and cerebellum. Damasio and Maurer [1978], focusing on autistic deficits in executive function, fingered the medial frontal lobes and the basal ganglia as sites of dysfunction. In their discussion of the medial frontal lobes they place particular emphasis on the role of the cingulate gyrus, an important part of the limbic system. This hypothesis was based on similarities between the behavior observed in people with autism and in adults with acquired medial frontal or basal ganglia lesions. A hypothesis put forth by Ornitz [1983] suggested that autistic difficulties with sensory modulation and with sensorimotor response could best be explained by a malfunction of brainstem and diencephalic networks that gate sensory input. Deficits in higher-order capacities such as language, he proposed, are consequences of the abnormal input supplied to the cerebrum by these malfunctioning gating systems. (As we shall see, this notion of a cascade of dysfunction, in which one malfunctioning brain system perturbs its functional consequents, is a recurring theme in theories of autistic neuropathology). Another possible explanation for the stimulus overselectivity manifested in autism is a developmental spatial neglect syndrome [Bryson & al. 1990], akin to the acute spatial neglect that follows parietal lobe injury. These authors reviewed a number of autonomic, electrophysiological and behavioral studies indicating the existence of attentional anomalies in people with autism and they describe similarities between the behavior of people with autism and that of patients with left-sided neglect. Hetzler and Griffin [1981] and DeLong [1992], recognizing the similarities between autistic behavior and Klüver-Bucy Syndrome, have characterised autism as the effect of developmental dysfunction in structures of the medial temporal lobe, especially the hippocampus and amygdala. This theory incorporates an observation that's sure to figure in any explanation of autism: the effect of a lesion in a developing brain often is more severe than or is distinct from the effect of a similar lesion in a mature brain, because brain systems downstream from the lesion may depend on correct input for their proper maturation. This is supported by Bachevalier's [1994] finding that lesions of medial temporal lobe structures induced in neonatal monkeys result in behaviors that are more typical of autism than similar lesions induced in adult monkeys.
As well as these anatomical systems, quite a few chemical systems have been thought to underlie autism. The finding of hyperserotonemia in some people with autism has been widely replicated [Cook 1990]. Levels of the other monoamines also have been found to be higher than normal in people with autism [Martineau & al. 1992]. Panksepp [1979], on the basis of autistic insensitivity to pain, suggested dysfunction of opiatergic systems, and others have confirmed elevated levels of opiate activity in cerebrospinal fluid [Gillberg & al. 1985] and urine [Gillberg & al. 1982; Gillberg 1988; Reichelt & al. 1991, Reichelt & al. 1994; Williams & al. 1991; Shattock & Lowdon 1991] of people with autism.
CEREBELLAR ANATOMY IN AUTISM
Each of these theories seems to contain at least a grain of truth, but how can they all be unified? Is there a single brain structure or system that could produce secondary malfunctions in all these other structures and systems? What region is consistently abnormal in autistic brains? So far, the most consistent neuropathological abnormalities have been found in an area which no one would have considered until recently: the cerebellum.
While the most noticeable and diagnostic characteristics of autism are cognitive in nature (e.g. impaired social interaction and communication, restricted range of interests, [American Psychiatric Association, 1992], a number of motor abnormalities have also been noted, although they receive relatively little attention clinically and scientifically. For instance, autistic children are universally dyspraxic [Jones & Prior 1985], and manifest disturbances of gait [Vilensky & al. 1981] which persist, to some extent, into adulthood [Hallett & al. 1993]. Motor anomalies are clear enough that Adrien & al. [1992] found that blind raters were able to differentiate autistic children from normal children on the basis of motility and other factors appearing in home movies taken before autism was suspected. In a more recent study using a battery of tests taken from a standard neurological exam, Haas & al. [1996] found that autistic patients showed abnormalities indicative of cerebellar and parietal lobe dysfunction, but did not show significant abnormalities on tests of pyramidal motor and cranial nerve abilities. Furthermore, classical eyeblink conditioning, a hallmark of cerebellar functioning, is abnormal in autism, both in the amplitude and latency of conditioned responses and in the rate of extinction [Sears & al. 1994].
The first indication of the neuropathological bases of these functional abnormalities came a decade and a half ago, when Williams & al. [1980] detected a low count of Purkinje cells in the cerebellum of one of four autistic brains at autopsy. Ritvo & al. [1986] confirmed this finding, reporting a 41% loss of Purkinje cells in the vermis and a 35% loss in the hemispheres in four autistic brains as compared to normal brains. In a more comprehensive study, Bauman [1991] reported depletion of Purkinje cells and granule cells in autistic brains at autopsy, with no evidence of the gliosis that ensues when neurons degenerate after infancy. In the cerebellar deep nuclei, the destinations of Purkinje cell axons, they found abnormally enlarged cells in autopsied children and a depletion of cells in autopsied adults. In the inferior olive, the source of climbing-fiber input to Purkinje cells, they found small cell bodies in the lateral portion, but no apparent loss of cells. This absence of retrograde cell death in the inferior olive, like the absence of gliosis in the cerebellar cortex, points to a time of onset early in fetal life. Bauman and Kemper [1993] later speculated that the olivary and deep nuclear abnormalities might be a consequence of the retention of a foetal pattern of cerebellar circuitry in which climbing fibers synapse directly on cells of the deep nuclei, without sending the major branches of their axons into the cerebellar cortex. Such a short-circuited pattern of connections would sabotage the regular topography of the cerebellar cortical map. The climbing fibers that do extend to the cerebellar cortex may innervate multiple Purkinje cells inappropriately, because of the depletion of granule cells which somehow mediate the formation of the correct, one-to-one or one-to-few mapping of climbing fibers to Purkinje cells [Crepel 1982].
By the late 1980s, nuclear magnetic resonance imaging studies began to reinforce the autopsy findings. The depletion of cells at the microscopic level manifested as a deficit in size at the macroscopic level, specifically, as a reduction in the cross-sectional area of the cerebellar vermis. Courchesne & al. [1987] reported this finding in a single case, and Gaffney & al. [1987] extended the finding to a group of eight subjects. In a larger study (18 autistic subjects), Courchesne & al [1988] localised this hypoplasia to vermal lobules VI and VII. Subsequent work in the same laboratory demonstrated that hypoplasia could be found not only in the vermis of the autistic cerebellum, but also in the larger cerebellar hemispheres [Murakami & al. 1989]. In the largest MRI study of the autistic cerebellum to date, Hashimoto & al. [1995] found hypoplasia of lobules VI and VII when a group of 102 people with autism were compared to 112 control subjects. This difference was found even in children less than 2 years old who were later diagnosed with autism. In contrast, studies from several other labs [e.g. Holttum & al. 1992], Hashimoto & al. 1993, Kleiman & al. 1992], using similar anatomical methods, failed to replicate these findings. There is a number of possible explanations for this discrepancy, including small sample size (none of the studies that obtained negative results used more than 18 autistic subjects), and the use of patient controls [Hashimoto & al. 1992, 1993, Kleiman & al. 1992] versus healthy volunteer controls [Courchesne & al. 1988, Piven & al. 1992, Holttum, & al. 1992, Garber & al. 1992].
It is also possible that non-specific IQ effects play a role in these findings. While some of the aforementioned studies chose control groups with IQs comparable to those in their autistic groups (e.g. Piven & al. 1992, Holttum, & al. 1992) most, including the studies that found significant differences in vermal area (Courchesne & al., 1988; Hashimoto et al., 1995), did not. It has therefore been suggested (Piven & Arndt, 1995; Filipek, 1995) that these differences were due to confounding differences in IQ rather than to a specific effect of autism. In fact, Schaefer et al. (1996) have presented evidence that a number of disorders can result in vermal hypoplasia, including disorders that do not show autistic-like behaviours. Needless to say, this is an important result and clearly emphasises the necessity of including IQ matched control groups in later studies. However, we must not forget that a single macroscopic difference (i.e. vermal hypoplasia) could result from a wide variety of distinct microscopic differences (e.g. loss of Purkinje cells in autism, loss of granule cells in Down's). Additional research is necessary to evaluate the specificity of both macroscopic and microscopic differences. And, we must keep in mind that even a non-specific finding gives us insight into the nature of the causal abnormalities of the autistic syndrome.
|
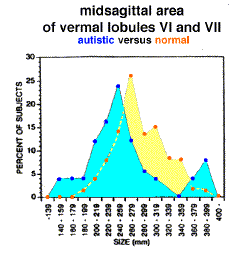
Another possible explanation for the discrepancies between studies that found significant differences and those that didn't was suggested when Courchesne & al. [1994] described the possibility of a hyperplastic subgroup in addition to the hypoplastic group previously described. Their analysis of 50 autistic subjects showed a bimodal distribution in the areas of lobules VI and VII with 5 of these subjects falling more than 2 standard deviations above the mean found in normal controls (fig. 1). If this result is verified in additional studies with large samples, this mix of subgroups would, as demonstrated by Courchesne & al. [1994ab], explain the inability of smaller studies to find significant differences between people with autism and control subjects.
While there has been a great deal of discussion [Courchesne & al. 1994, Piven & Arndt 1995, Filipek 1995, Schaefer & al. 1996] as to whether MR studies demonstrate hypoplasia of the vermis in the autistic cerebellum and whether this effect is specific to the disorder, an appraisal of all the available data indicates that cerebellar abnormalities are an important aspect of autism. This includes both the neuropathological studies, all of which have found cerebellar abnormalities especially of the Purkinje cells, and the neuroimaging studies, in which those with the largest sample sizes (and therefore the greatest statistical power) have found cerebellar hypoplasia.
It's important to note here that the cerebellar abnormality is not limited to the vermis. In reviews of this topic, the earlier work on the vermis often is cited without mention of the hemispheric finding (i.e. Murakami & al. [1989]), an unfortunate omission since it leads people to believe that the hemispheres are unaffected. In fact, the hemispheres are almost as greatly affected as the vermis. The only reason the vermis has received the majority of research is that it is technically easier to measure: its anatomy provides a ready plane for the alignment of MRI slices such that any alignment error is immediately discernible from alteration of midsagittal cerebellar landmarks, and the landmarks are sufficiently obvious to make the outlining of the area a straightforward process. However, the evidence of decreased size of the hemispheres does also exist and future research and reviews should emphasise the full anatomical extent of cerebellar pathology in autism to give a more accurate perspective.
THE CEREBELLUM AND COGNITION
It's easier to understand how cerebellar damage might produce autistic cognition and behavior if we first understand the role of the normal cerebellum in normal cognition. Leiner, Leiner, and Dow [1986, 1991] were the first to argue forcefully for the involvement of the cerebellum in non-motor operations. They based their conjecture on the concomitant evolution of the prefrontal cortex and the neocerebellum, and on the idea that a single computational architecture could implement both motor and non-motor representations. These influences could be achieved through the neuronal connections that exist between the cerebellum and multiple regions of the cerebrum. In monkeys, the cerebellum projects via the thalamus to the prefrontal cortex [Middleton & Strick 1994]. In cats, the cerebellum projects to the hippocampus via the thalamus, and appropriately timed stimulation of the cerebellar vermis potentiates hippocampal response to somatosensory input [Newman & Reza 1979]. In rats, stimulation of the vermis modulates responses of the brainstem, the tectum, and the cerebral cortex to visual, auditory, and somatosensory stimuli [Crispino & Bullock 1984]. In humans, cerebellar strokes can result in decreased metabolism in prefrontal and temporal cortices [Attig & al. 1991]. In short, the cerebellum is ideally situated to influence absolutely all of the cognitive systems and anatomical regions that have been implicated in all of the various theories of autism.
Several functional imaging studies have carried this relationship a step further, by going beyond the static relationships of anatomy and examining the functional relationship between the cerebellum and the rest of the brain. Petersen & al. [1989] showed that the right lateral cerebellum is active during a semantic language task, but not during a language repetition task. Specifically, the cerebellum was active when subjects had to produce spoken verbs whose meanings were related to nouns that they read, but not when the subjects had merely to speak the nouns. With practice, however, this cerebellar activation, along with prefrontal activation in the cerebral cortex, diminishes [Raichle & al. 1994]. Especially in light of the anatomical projection of the cerebellum to prefrontal cortex, this suggests that a cerebello-thalamic-prefrontal control system is involved in the creation of novel cognitive strategies, but not for rote execution of such strategies. Similarly, in subjects attempting to solve a pegboard puzzle, activation of the dentate nuclei was three to four times larger than the activation that occurred during simple, visually guided movements of the pegs [Kim & al. 1994]. In tasks requiring tactile discrimination, the dentate nucleus again was much more active than in cases of similar but passive palpation [Gao & al. 1996]. In a visual `oddball' task, which required a simple visual discrimination without a motor response, the superior-posterior cerebellar cortex was strongly activated, but this area was only very weakly activated by a simple motor task. Similarly, when the discrimination task was combined with a contingent motor response, the activation was no greater than in the non-motor discrimination task [Allen & al. 1997]. On the basis of these first few studies, the role of the cerebellum in cognition might be characterised in general as the adaptive selection of cognitive strategies. Functional scanning of the cerebellum hasn't yet been attempted in people with autism, probably because these subjects often have difficulty lying still in the scanner and because even the normal cerebellar contribution to cognition is only just beginning to be characterised. We can look forward, though, to a growing understanding of cerebellar functioning, and the beginning of an inquiry into the specifics of its abnormal functioning in autism. Functional imaging techniques will be critical to the development of this understanding.
An important point to emphasise when discussing the role of the cerebellum in autism is the distinction between early developmental cerebellar malformation and other types of cerebellar abnormality. It might be argued that the cerebellum can't be responsible for autism, since people whose cerebella become damaged or are congenitally absent don't behave anything like people with autism. This argument overlooks the developmental nature of the deficit. Time and time again in the study of neural systems, we find that appropriately structured input is required for correct development. In the cerebellum, the Purkinje cells of the cerebellar cortex are the sole inhibitory input to the deep cerebellar nuclei. Loss of appropriately patterned inhibition could produce constitutive activation of the deep nuclei, and, consequently, constitutive, nonspecific activation downstream in the cerebral cortex. Such a situation may very well be worse for development than having no cerebellum at all! Even aside from these biological considerations that may make congenital or early developmental defects more damaging than acquired defects, there may be psychological considerations as well. How might congenital impairment of the attentional and other cognitive functions of the cerebellum differ from acquired impairment in the nature and scope of its effect?
The development of joint social attention is a major milestone in the maturation of a normal child. It marks the beginning of a baby's ability to learn the nature of its world under the guidance of its parents and other adults. The learning process is thus greatly accelerated. A properly functioning attentional system is essential for the timely development of joint social attention, and on joint social attention in turn depends the explosion of learning that characterises every normal toddler. Imagine the following scene. A baby rests on his father's lap. Outside a window, a flash of sunlight glints from a fender, a car horn sounds, there is a crunch of gravel, and the throb of an engine grows and then abruptly ceases. The father strokes the baby, smiles at him, and announces that the baby's mother has arrived. The father has made an inference about the world, and a normal baby would learn eventually to make the same inference when presented with the same conjunction of stimuli. Implicit in this process is a tremendous degree of integration of sensory stimuli: the father's gentle touch, his tone of voice, the particular words that he speaks, his smile, all figure in the representation of mother; the glint of light, the sounds of the engine, of the horn, and of the gravel beneath the tires all point to the fact that a car is entering the driveway. Combine this with the stored knowledge that mother has been out in a car, and the inference is made. But if the baby has difficulty with attentional shifting or distribution, then only one or a few of these many stimuli may reach awareness. They come in rapid sequence, via various sensory modalities, and from diverse locations in space. Without a properly functioning mechanism for rapidly and automatically shifting and distributing attention, the task of integrating all of them into a coherent percept is rendered very difficult, to say the least. In a developing infant, who has no prior knowledge of the world, learning may become almost impossible. Thus damage to olivoponto-cerebello-thalamocortical control loops that optimise attentional dynamics, though a rather innocuous deficit in itself, may begin a cascade of detrimental effects that results in profound alteration of cognitive and social development.
|
If this is the case, then it should be possible to observe the primary attentional difficulty in brain-injured adults, without the profound, secondary, developmental effects that result from the congenital form of such damage. Akshoomoff and Courchesne [1992] have observed just such a dissociation. Their experiment consists of two `oddball' paradigms running simultaneously. In the standard oddball paradigm, there are two types of stimuli, a relatively rare, target stimulus, and a common, background stimulus. The experimental subject must produce a motor or mental response to each instance of the target, but not to the background stimuli. In a visual oddball experiment, for example, the target stimulus might be a red flash that occurs with 20% probability, and the background stimulus might be a green flash occurring with 80% probability. In an auditory oddball experiment, high and low tones might be used for the same purposes. In the attentional-shifting experiment, visual and auditory oddball stimuli are presented concurrently, and subjects must attend one modality, either vision or audition, while ignoring the other. A target stimulus in the attended modality cues not only an overt motor response but also a covert shift of attention to the previously unattended modality. The next target in this newly attended modality cues a shift back to the other modality, and so on. Akshoomoff's original experiment was a behavioral measure of the execution time of attentional shifts between visual and auditory modalities. She found that patients with cerebellar damage were impaired specifically in detecting targets that occurred in a newly attended modality within the first few seconds following the cue to shift from the previously attended modality. Later, this result was replicated in people with autism [Courchesne & al. 1994], and in tasks that required division of attention between auditory and visual modalities and between several attributes of visual stimuli [Casey & al. 1993]. (This latter study, however, used a heterogeneous group of subjects who had either autism or some other pervasive developmental disorder, and who had unusual calendar-calculating ability.) A second version of the shift experiment [Akshoomoff & Courchesne 1994] tested intramodality shifts of attention, in which the attended features were the color and the form of visual stimuli presented at a single spatial location. A similar behavioral result was found in this study.
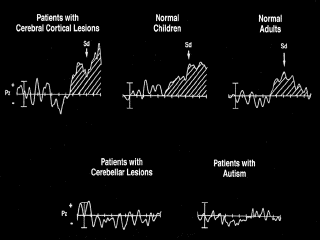
Reinforcing this behavioral result was an absence, in cerebellar patients and in people with autism, of the positive shift-difference (Sd) potential which in normal brains appears with a long latency (~700ms) after a cue to shift attention (fig. 2). This difference potential may be akin to the Late Directing Attention Positivity (LDAP) reported by Harter [Harter & al. 1989, Harter & Anllo-Vento 1991] in another attention-shifting experiment. Rather than using a continuous back-and-forth shifting paradigm, this experiment examined ERPs in response to an attention-directing cue in the center of the visual field. A cue to shift attention was followed, in the hemisphere contralateral to the cued side, by an occipitoparietal negativity at about 200ms, and then by the LDAP from about 400ms to 700ms. These effects arose in difference waves comparing responses at each electrode to left cues and to right cues.
|
In an experiment based on the spatial attentional cueing paradigm of Posner [1980], Townsend found in cerebellar patients and in people with autism the same behavioral deficit for short cue-to-target intervals as had been found in the previous intermodality and intramodality studies (fig. 3) [Townsend & al. 1996a, b]. That is, people with autism and cerebellar lesion patients are abnormally slow to shift attention in space or between or within sensory modalities. The behavioral and electrophysiological effects shown by cerebellar patients and by people with autism in these experiments which address selectively the capacity for rapid shifting of attention support the hypothesis that cerebellar damage in autism produces a deficit in such shifting, which may further impair cognitive and social development.
TEMPORAL LOBE
The cerebellum seems the most consistently abnormal structure in the autistic brain, and it would simplify matters if the cerebellum were the only abnormal structure. But the picture isn't so straightforward. In the same studies in which they noted depletion of cerebellar granule cells and Purkinje cells, Bauman and Kemper [1985] also reported pathological changes in cell morphology in limbic structures. In the hippocampus, amygdala, anterior cingulate, entorhinal cortex and mamillary bodies they noted small, closely packed cells. A Golgi analysis of two subjects [Raymond, Bauman & Kemper 1989] also showed a decrease in dendritic branching of CA1 and CA4 hippocampal neurons. They speculated that these changes, like those in the cerebellum, may reflect incomplete development [Bauman & Kemper 1993].
Additional evidence supporting a role of temporal lobe dysfunction in autism comes from epilepsy research. Epilepsy is believed to be relatively common in people with autism (as high as 25%, American Psychiatric Association, 1994), and some forms of temporal lobe epilepsy with onset in early childhood can mirror at least some of the symptoms of autism. Landau-Kleffner Syndrome [Landau & Kleffner 1957; Deonna 1991], for example, involves a malformation of the temporal lobe which results in a loss of language as well as seizures. Since a seizure focus in the temporal lobe can mimic autistic symptoms, then it seems logical to hypothesise that autism, at least in some of its forms, involves abnormal function of the temporal lobe. Reinforcing this hypothesis are several case reports of autistic behaviors acquired in older children [DeLong & al. 1981; C. Gillberg 1986], and even in one adult [I.C. Gillberg 1981] following acute encephalitis. In each of these cases an autism-like syndrome developed rapidly (1-3 months) in previously normal people and was presumed to be due to temporal lobe dysfunction as evidenced by abnormal temporal lobe EEG and, in two cases, CT or MRI. (It is interesting to note that two of these patients recovered fully from the autistic syndrome [DeLong & al. 1981]). Another study [Hoon & Reiss 1992] described a fairly classic case of autism (first signs at 1 to 2 years) which was associated with a unilateral temporal lobe tumor and seizures.
Another condition that seems to have some symptoms in common with autism is the Klüver-Bucy Syndrome. This syndrome has primarily been described in monkeys with temporal lobe lesions. It occurs in humans only in rare cases following bilateral lesions of the temporal lobe due to encephalitis, or, in past instances, to psychosurgery. The symptoms are a severe visual agnosia, also known as `psychic blindness', nonresponsiveness in social contexts, flatness of affect, and a tendency for oral and somatosensory exploration with relative disuse of vision and hearing. Bachevalier [1994] described a related but distinct disorder in monkeys which received temporal lobe lesions neonatally rather than in adulthood. At two months of age, these monkeys showed increased passivity and a decreased tendency to initiate social contact when compared to normal monkeys. By six months of age the same monkeys showed an even greater degree of abnormality by actively avoiding social contact and demonstrating locomotor stereotypies. These monkeys did not demonstrate hypersexuality, a symptom of the Klüver-Bucy syndrome which is not seen in autism, nor did they demonstrate the loss of fear that is characteristic of Klüver-Bucy.
While these diverse cases offer insight into the possible bases of autistic symptoms, imaging studies are in a unique position to point us to a correct localization of the physiological abnormalities in classic autism. A detailed imaging study [Saitoh & al. 1995] has shown no macroscopic decrease in cross-sectional area of the posterior hippocampus in people with autism as compared to normals. The temporal lobe as a whole, in fact, is larger in people with autism than in normals [Osamu Saitoh, unpublished observations]. These somewhat paradoxical findings illustrate the difficulty of mapping macroscopic imaging data onto microscopic, histoanatomical observations. Nevertheless, on the basis of the neuropathological data, it does seem that some process of abnormal development is occurring in the medial temporal lobe of the autistic brain. The neuronal connections between the cerebellum and the temporal lobe raise the possibility that temporal and limbic maldevelopment may, at least in some cases, be a consequence of cerebellar maldevelopment. Thus there is no shortage of possible mechanisms for the unification of the limbic theory of autism with the cerebellar theory.
Much more imaging work on the temporal lobe in autism remains to be accomplished. The posterior hippocampus is the only individual structure of the medial temporal lobe that has so far been investigated quantitatively using MRI. More anterior regions such as the amygdala remain unexamined. Functional imaging also has much more to contribute to our knowledge of the temporal lobe in autism. To date, SPECT studies [George & al. 1992; Mountz & al. 1995] have demonstrated decreased perfusion of the temporal lobe, but these studies used small samples. Furthermore, the progression of this abnormality over the course of development remains unclear.
PARIETAL LOBE
The list of symptoms described above in the context of Klüver-Bucy Syndrome seems, at least superficially, to describe autism. Of course, symptoms of agnosia can to a large extent be mimicked by symptoms of attentional neglect. Furthermore, the social and emotional abnormalities of autism may be developmental epiphenomena of such a lack of attentional control. So autism might be explained by either, or both, or neither of the temporal (gnostic) theory and the parietal (attentional) theory. Hetzler and Griffin [1981] characterised autism as a deficit in the formation of cross-modal associations. Here again, though, the question is one of primacy: is there some defect in the associative processing that binds multimodal sensations into percepts, or is the apparent associational deficit a product of a more fundamental lack of ability to distribute or to shift attention among several sensory modalities simultaneously?
Bryson & al. [1990], as mentioned at the beginning of this chapter, hypothesised that autism is a developmental neglect syndrome, related to the hemispheric neglect found in patients with parietal lesions. There is imaging evidence to support this theory, at least in some cases. Forty-three percent of people with autism in the subject group of Courchesne & al. [1993] had parietal sulcal widening, as determined in a blind assessment by a neuroradiologist. If autism is a developmental version of parietal lobe lesions, then the developmental version is much more pervasive than the acquired version, just as in the case of developmental cerebellar pathology. This would be due to downstream consequences of the deficit to the development of other brain regions and because of the absence of fully matured, alternative cognitive faculties which might be used to compensate for a deficit in one particular aspect of cognition. It is generally believed that the parietal cortex, especially the right hemisphere, is important in spatial attention [Heilman & al. 1985]. While the parietal anatomical deficit in autism has been assessed qualitatively, it still awaits quantitative study which may reveal decreased parietal lobe volume in a larger proportion of autistic brains.
Reinforcing the finding of a deficit in parietal tissue volume is the finding of narrowing in the posterior corpus callosum [Egaas & al. 1995], the region of the callosum that contains fibres crossing between the parietal lobes [Pandya & al. 1971]. An MRI study of callosal myelination in autism [Belmonte & al. 1995] detected no difference between people with autism and normals, even though its methods were sensitive enough that they detected an age-related progression in myelination. This suggests that the axons that are present are myelinated to a normal extent, and that the deficiency in cross-sectional area must result from a deficiency in the number of axons. Putting two and two together, it seems as if the normal complement of layer V pyramidal cells may be absent from the parietal lobes in at least some cases of autism. This hypothesis awaits histoanatomical study.
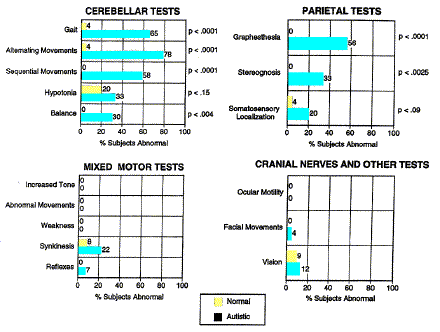
The parietal deficit detected by MRI is also corroborated by behavioral and electrophysiological studies. In the neurological examination by Haas & al. [1996] mentioned previously, the only category of tests other than cerebellar shown to be abnormal in people with autism was the group of parietal tests assessing graphesthesia, stereognosis, and somatosensory localization (fig. 4). In Posner's [1980] attention-shifting paradigm, people with autism demonstrate a heightened validity effect at long cue-to-target intervals [Casey & al. 1993, Wainwright-Sharp & Bryson 1993]. That is, they take longer than normal to respond to a target at an unpredicted location when their attention has already been engaged at an erroneously predicted location. Townsend & al. [1996] have shown that this heightened validity effect is due specifically to the subgroup who have parietal sulcal widening, and that in fact the degree of the behavioral effect correlates with parietal volume loss.
In the normal brain, attention to the spatial location of a stimulus operates by a mechanism of `early selection', that is, all stimuli at an attended location, whether their particular features are relevant or irrelevant, enhance neural response even at relatively short latencies [Heinze & al. 1990]. The competing theory, that of late selection, would predict a dependence of brain response to stimuli at an attended location on whether or not the stimuli possessed attended features. So, for example, if an experimental subject were instructed to respond to a square in the upper left corner of the screen, her brain electrical response would be stronger to that target stimulus than to a circle appearing in the same location. In fact, during the first few hundreds of milliseconds following the stimulus, this isn't what's observed: the amplitude of the occipitotemporal P1 evoked potential is increased in response to any stimulus in the upper left corner, not just to the square. These data suggest that spatial attention operates independently of, and in advance of, attention to features. Such a computational architecture makes sense, because the extraction of features is a much more complex procedure than the simple identification of the appearance of a new stimulus at a particular location.
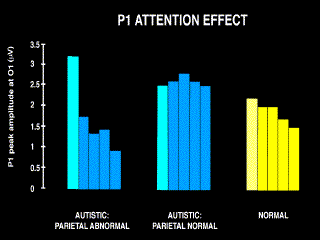
Attentional enhancement, instead of being organised in a `spotlight' with distinct spatial boundaries, operates as a spatial gradient centered on the attended locus. For example, P1 amplitude is greatest for stimuli at the attended location, but is nearly as great in response to stimuli occurring close by the attended location. Rather than a definite cutoff, there is a gradual decline in amplitude with increasing eccentricity from the focus of attention [Mangun & Hillyard 1987, 1988]. The close functional association of the P1 with attentional processing is further supported by the fact that behavioral measures of accuracy, such as detectability and reaction time, covary with P1 amplitude [Mangun & Hillyard 1990]. In people with autism, though, the story differs. They distribute visual spatial attention abnormally, even in conditions of static attention without any requirement to shift from one location to another. In an experiment by Townsend and Courchesne [1994], autistic subjects were presented with brief flashes, each at one of five separate locations. Subjects were asked to respond with a button press to flashes in one of the locations, but to ignore flashes in the other locations. Normal subjects responded with a spatial gradient of P1 amplitude enhancement. A subgroup of autistic subjects that had parietal sulcal widening visible on MRI scans operated more in keeping with the spotlight metaphor; their P1 in response to stimuli at the attended location was actually larger than normal, but their P1 in response to stimuli even slightly eccentric to the attended location was much smaller than normal. This suggests an abnormally focused allocation of attentional resources to one particular point in space, with a relative neglect for the surrounding space. In contrast, autistic patients without obvious parietal volume reduction evidenced little enhancement of P1 to stimuli presented at an attended location as compared to the P1 to stimuli presented at surrounding locations (fig. 5). (This latter result, however, still needs to be verified with a larger group of parietally normal autistic subjects. Work on this is in progress.) It's typical of the pervasive nature of autism that a defect in one brain area, the parietal lobe, is best observed electrophysiologically by examining responses generated in another area, the occipital lobe. By observing the perturbed functioning of the visual system, it's possible to trace the dysfunction back to the attentional system.
|
The P1 experiment tells us a lot about dysfunction in statically allocated attention, but it can't address the neurophysiology of shifts in attention. Shifts in attention are difficult to assay using conventional event-related potentials, because the latencies and durations of sensory evoked responses are in general comparable to--or even greater than--the time courses of attentional shifts. Investigating such rapid attentional processes using such relatively slow signals would be like trying to repair a watch with a sledgehammer. In order to investigate attentional dynamics using evoked responses, we've adopted the paradigm of the steady-state visual evoked potential (SSVEP). The SSVEP is an electrical resonance in the visual system that arises in response to stimuli flashed at regular intervals. Stimulus frequencies in the alpha band (about 8s-1 to 12s-1), the brain's natural resonance frequency, produce particularly powerful SSVEPs. In normal brains, allocation of spatial attention to a particular location modulates the brain responses in the occipitotemporal region of the hemisphere contralateral to the newly attended stimulus. In particular, the amplitude of the SSVEP is augmented, while the amplitude of background activity which is not phase-locked to the flashing stimulus is diminished [Belmonte 1998a]. These differentials can be detected using specialised algorithms [Belmonte 1997], and changes can be observed as the subject shifts attention from one flashing stimulus to another at a different location.
The autistic brain, in contrast, manifests no such modulation. Rather, both the SSVEP and the background responses in both hemispheres are greater in response to stimuli in the left visual field than in response to stimuli in the right visual field [Belmonte 2000]. This tendency does manifest in the normal brain, in that attentional augmentation of right-hemisphere responses to left-field stimuli is in general greater than attentional augmentation of left-hemisphere responses to right-field stimuli. It seems that what's missing in the autistic brain is the hemispheric specificity. It's quite possible--and, we think, probable--that this lack of hemispheric specificity is just the most obvious manifestation of a general lack of spatiotopy in attentional modulation of brain responses. This certainly would agree with the lack of spatially specific P1 enhancement described by Townsend and Courchesne [1994].
FRONTAL LOBE
The frontal lobe has yet to be addressed quantitatively by anatomical imaging studies, but results of functional imaging and event-related potential studies do suggest its involvement. Much has been made of the so-called `theory of mind' deficit in autism [Baron-Cohen 1989], and frontal dysfunction may underlie a specific deficit in making the abstract inferences about the social world that are often described as the essence of a theory of mind. In normal subjects, orbitofrontal cortex seems selectively activated during a task involving theory of mind [Baron-Cohen & al. 1994], but it remains unclear whether this effect reflects specifically the second-order social inference characteristic of theory-of-mind tasks, or whether it reflects a more general difference in task complexity.
Ozonoff & al. [1991] hypothesise that the apparent deficits in theory of mind may be secondary to a general deficit in executive function. Such deficits are more universal among people with autism than are deficits in theory of mind. Perhaps people with autism are more impaired at applying a theory of mind than at constructing one: they may be able to understand the theory-of-mind task, yet be unable to avoid responding in terms of what they themselves know rather than in terms of the knowledge that they attribute to others. There does seem evidence for a more general deficit in executive dysfunction in autism. In a task that was similar to the Wisconsin card sorting test but whose changes of sorting criterion involved either a change in what feature was relevant or a reversal of the relevant value of the relevant feature, people with autism were selectively impaired in responding appropriately to the changes between features [Hughes & al. 1994]. This deficit in executive function is reminiscent of findings of stimulus overselectivity in autism [Lovaas & al. 1971], and indeed, stimulus overselectivity on the input side may contribute to perseverative classification on the output side.
The normal frontal negativities associated with auditory attention and with visual attention are absent in autism [Courchesne & al. 1985, Ciesielski & al. 1990]. (People with autism also manifest a reduced auditory P3 component [Novick & al. 1979, 1980; Courchesne & al. 1984, 1985, 1989], and some of this reduction results from an increase in variability of the response: in single trials it is sometimes present, sometimes not.) Interestingly, the behavioral performance of people with autism in vigilance tasks that do not involve shifts of attention is normal, despite the lack of these potentials. Either the P3 and the Nc don't reflect processes that are essential to behavior, or people with autism develop some compensatory strategy that yields the same appropriate behavior but used alternative brain mechanisms.
A functional imaging study has added to the evoked-potential evidence of frontal dysfunction in autism. A longitudinal study using SPECT [Zilbovicius & al. 1995] revealed an apparent developmental retardation of the frontal cortex. Three-year-old autistic children showed low levels of frontal cerebral blood flow, resembling the levels of normal one-year-olds more than those of normal three-year-olds. By six years of age, however, frontal blood flow in the autistic children had normalised. Normal frontal function in the intervening years may be critical for normal social development, though, so this transient dysfunction could do permanent damage.
OTHER NEUROANATOMICAL FINDINGS
In addition to the diverse neuroanatomical findings already described, a few more general observations have been made with regard to the autistic brain. Piven & al. [1995] reported that total brain tissue volume in people with autism was significantly larger than that of normal control subjects based on MR images. This result agreed with a previous study from the same laboratory [Piven & al. 1992] which had found a larger mid-sagittal brain area in people with autism than in controls and with an abstract from Filipek & al. [1992] which reported an increased volume. Hopefully, additional research will be done to determine if these differences are due to localised differences in brain size (e.g. specific to a particular lobe or structure) or if they represent a diffuse difference throughout the brain.
Another study from the Piven laboratory [Piven & al. 1990] examined MR images of autistic and control brains for evidence of gyral malformation. Scans were assessed for the presence of abnormalities such as polymicrogyria, pachygyria, heterotopia and schizenchephaly which the authors believe result from defects in neuronal migration. They found one or more such malformations in 7 of 13 autistic subjects but in none of the 13 control subjects. The malformations were not specific to any particular lobe or side of the brain nor did they all fall into a single category of malformation. This suggests that defects in cortical neuronal migration may play a role in some cases of autism and that such defects may have multiple ætiologies. Again, further research with additional subjects should shed light on the frequency of the various defects and possibly on the root causes of the abnormalities.
BIOCHEMICAL BASES OF IMAGING RESULTS
So much is wrong in autism that it's difficult to single out a primary dysfunction. One candidate, cerebellar maldevelopment, may well be the explanation for many cases. But it would be naïve to suggest that the cerebellum might be the be-all and the end-all of autistic disorder. Undoubtedly there are multiple ætiologies at work, and these will be difficult to disentangle. Furthermore, the cerebellar explanation itself demands its own explanation: if the cerebellum is developing abnormally, if Purkinje cells are dying, or failing to differentiate or to migrate, then what cellular and molecular mechanisms are responsible?
As mentioned in the introduction to this chapter, the cerebrospinal fluid and urine of many people with autism have high levels of opiate binding. Reichelt & al. [1991, 1994], and also Shattock and his colleagues [Williams & al. 1991; Shattock & Lowdon 1991], have suggested that these opioids may result from incomplete digestion of gluten or casein and consequent passage of opioid peptides from the gut into the circulation, and thence across the blood-brain barrier. Whatever the source of these opioids, they are definitely present in the central nervous system in many people with autism. Since opioids act during development as inhibitory neuronal growth factors, their presence to excess in the developing brain could produce the very cellular effects that have been noted in the autopsy studies discussed above. Morphine in low concentrations reduces dendritic arborization in murine Purkinje cells in vitro, and in higher concentrations kills these cells [Hauser & al. 1994]. In normally developing cerebellar cortex in the rat, messenger RNA for proenkephalin is expressed in Purkinje cells, in spatiotemporal gradients that match regional rates of maturation [Osborne & al. 1991, 1993]. The enkephalins themselves are concentrated in the external germinal layer, where neural stem cells proliferate and differentiate [Zagon & al. 1985, 1987]. This suggests a process of opioid-mediated signalling in proliferation or differentiation. The human [zeta] opioid receptor subtype is expressed in the cerebellum only in infants [Zagon & al. 1990], so it may be the most important in this process. Large, daily doses of the opioid antagonist naltrexone, sufficient to block opioid receptors continuously throughout postnatal development, result in cerebellar hyperplasia. Low doses of naltrexone, which block opioid receptors only briefly and so produce an overall up-regulation of opiatergic transmission, result in cerebellar hypoplasia [Zagon & McLaughlin 1986a, 1986b]. This paradoxical effect suggests a tantalizing link to the bimodal distribution of cerebellar sizes in autistic brains. The same, bi-directional effect occurs when Leu-enkephalin is added to cultured rat raphé cells [Davila-Garcia & Azmitia 1989]. In addition to their cerebellar effects, opioids distort the development of the cerebrum. The same low dose that produces cerebellar hypoplasia also produces increased cell packing density in the cerebrum, particularly in the hippocampus. Recall that such increased packing is exactly what Bauman and Kemper [1985] have reported in some autopsied autistic brains.
In addition, genetic factors are certain to play a modifying or contributing part in many cases of autism. This much is handily demonstrated by the great difference in concordance for autism between monozygotic and dizygotic twin pairs [36% of 11 pairs vs. 0% of 10 pairs respectively, Folstein & Rutter 1977], and by the clustering of affective disorders in relatives of people with autism [DeLong & Nohria 1994]. The hyperserotonemia common in autism occurs also in relatives of people with autism [Cook 1990], and in fact correlates with levels of depressive and obsessive-compulsive symptoms [Cook & al. 1994]. In a significant step toward a possible molecular genetic basis for this tendency, Comings & al. [1991] reported that the incidence of the A1 allele of the dopamine D2 receptor gene in the autistic population is double its incidence in controls. A peculiarity of the D2 receptor gene may be associated in particular with the regressive subtype of autism, in which children begin to develop normally but then lose language and social abilities [Alan Lincoln, unpublished observations]. Reiss & al. [1986] have noted that the site of CGG repeats in Fragile X Syndrome, in which probands have some autistic features, is close to the location of several genes involved in cerebellar development. Daniels & al. [1995] reported association of autism with C4B null allele of the major histocompatibility complex, suggesting a genetic basis for the immune abnormalities observed in autism.
None of these candidate modifying factors are mutually exclusive. Genetic liability may magnify the effects of damage due to opioids or to malfunction of the immune system. The opiatergic and aminergic neurotransmitter systems interact strongly with one another, and can influence the immune system [Marchetti & al. 1990].
FUTURE DIRECTIONS
Over the last two decades a number of theories, both anatomical and chemical, have been presented to explain the biological bases of autism. All of the arguments come with some supporting evidence and some conflicting, but none is yet conclusive. Of the data accumulated so far, the most extensive and consistent relate to the cerebellum and the limbic system. Theories regarding these structures have support from neuropathological, anatomical, physiological and behavioral studies. Discussion continues, however, and more studies must be done before we can draw any concrete conclusions.
A complete understanding of autism will lead not only to prophylaxis for future generations but also to more effective treatments and coping strategies for people who currently have autism. This understanding can arise only from a synthesis of developmental neuroanatomy and neurophysiology with biochemistry and molecular genetics. While a comprehensive understanding of autism will require knowledge of the cellular and molecular abnormalities involved in the disorder, this level of understanding is likely to be a long time off. Research into these areas generally requires an animal model, which does not yet exist, or autopsy material, which is difficult to obtain. Since the disorder of autism does not affect longevity and longitudinal studies of autism are fairly recent and tend to recruit younger subjects, very little autopsy material is available for neuropathologic research. Because of this, in vivo neuroimaging techniques are absolutely vital to autism research. These techniques allow us to examine the integrity of brain structures at the macroscopic level and thereby infer microscopic anomalies, and as the technology evolves, will allow us to investigate smaller and smaller structures.
Future neuroimaging studies will ask such questions as: What is the developmental time course of the process of neurological damage that leads to autistic behavior? Since the diagnosis of autism can be made with certainty only when the child in question has reached the age of five years, there's a dearth of imaging data on very young autistic children. These early years, however, are precisely the period during which the most interesting anatomical and physiological developments (or maldevelopments) must be occurring. A study in progress in the Courchesne laboratory aims to fill this gap, by imaging tentatively diagnosed children at very young ages, and then confirming the diagnosis at the age of five years (an approach similar to that used by the Hashimoto laboratory). Without the proliferation of imaging technology, the luxury of over-inclusive scanning followed by retrospective diagnosis would be impossible.
What brain structures are affected in autism, how much are they affected, in what subpopulations, and over what time courses? While the cerebellum has received a great deal of attention in this regard, other important candidate areas have still received very little study due to the recency of an extensive interest in the biological bases of autism and of the advent of MR technology. Quantitative imaging studies of parietal and frontal lobes and additional limbic structures, in particular, are attractive prospects, because of the association of these structures with attention, executive function and social function. Such quantitative techniques must be combined with longitudinal studies in order to elucidate the time courses of abnormalities in various anatomical regions, and thereby to gain insights on which anatomical defects may be primary and which may be later, downstream effects. Automated analyses of MRI scan data will be essential to such quantitative studies and are currently under development.
Imaging allows us to examine macroscopic structure, to peer from the top down at the perturbed brain development that manifests as autism. Other, complementary technologies allow us to examine autistic development from the bottom up. A synthesis of both views is critical for progress toward a complete understanding of the autistic syndrome. What are the cellular and molecular mechanisms that contribute to the anatomical abnormalities seen with MRI? How might neuroanatomical and neurophysiological subtypes (cerebellar hypoplastic vs. cerebellar hyperplastic, parietally damaged vs. parietally normal) relate to ætiological, biochemical, and genetic subtypes (behaviorally regressive vs. non-regressive, viral vs. non-viral, hyperserotonemic, opioiduric, &c.)? Much progress has been made, but many new, more specific questions have been generated.
REFERENCES
Adrien JL , Perrot A, Sauvage D, Leddet E, Larmande C, Hameury L, Barthélémy C (1992) Early symptoms in autism from family home movies. Evaluation and comparison between 1st and 2nd year of life using I.B.S.E. scale. Acta Pædopsychiatrica 55:2:71-75
Akshoomoff NA, Courchesne E (1992) A new role for the cerebellum in cognitive operations. Behavioral Neuroscience 106:5:731-738
Akshoomoff NA, Courchesne E (1994) ERP evidence for a shifting attention deficit in patients with damage to the cerebellum. Journal of Cognitive Neuroscience 6:4:388-399
American Psychiatric Association (1994) Diagnostic and statistical manual of mental disorders, 4th edition. American Psychiatric Association, Washington, DC
Attig E, Botez MI, Hublet C, Vervonck C, Jacquy J, Capon A (1991) Diaschisis cérébral croisé par lésion cérébelleuse: Rôle du cervelet dans les fonctions mentales. Revue Neurologique 147:3:200-207
Bachevalier J (1994) Medial temporal lobe structures and autism: A review of clinical and experimental findings. Neuropsychologia 32:6:627-648
Baron-Cohen S (1989) The autistic child's theory of mind: A case of specific developmental delay. Journal of Child Psychology and Psychiatry 30:2:285-297
Baron-Cohen S, Ring H, Moriarty J, Schmitz B, Costa D, Ell P (1994) Recognition of Mental State Terms. Clinical findings in children with autism and a functional neuroimaging study of normal adults. British Journal of Psychiatry 165:5:640-649
Bauman ML (1991) Microscopic neuroanatomic abnormalities in autism. Pediatrics 87:5:791-796
Bauman ML, Kemper TL (1985) Histoanatomic observations of the brain in early infantile autism. Neurology 35:6:866-874
Bauman ML, Kemper TL (1993) The Contribution of Neuropathologic Studies to the Understanding of Autism. Neurologic Clinics 11:1:175-187
Bryson SE, Wainwright-Sharp JA, Smith IM (1990) Autism: A developmental spatial neglect syndrome? In: Enns JT (ed) The development of attention: Research and theory. Elsevier, Amsterdam, p 405
Casey BJ, Gordon CT, Mannheim GB, Rumsey JM (1993) Dysfunctional attention in autistic savants. Journal of Clinical and Experimental Neuropsychology 15:6:933-946
Cook EH (1990) Autism: Review of neurochemical investigation. Synapse 6:3:292-308
Cook EH , Charak DA, Arida J, Spohn JA, Roizen NJ, Leventhal BL (1994) Depressive and obsessive-compulsive symptoms in hyperserotonemic parents of children with autistic disorder. Psychiatry Research 52:1:25-33
Ciesielski KT, Courchesne E, Elmasian R (1990) Effects of focused selective attention tasks on event-related potentials in autistic and normal individuals. Electroencephalography and Clinical Neurophysiology 75:3:207-220 [erratum in 76:6:566 (1990)].
Comings DE, Comings BG, Muhleman D, Dietz G, Shahbahrami B, Tast D, Knell E, Kocsis P, Baumgarten R, Kovacs BW, Levy DL, Smith M, Borison RL, Evans DD, Klein DN, MacMurray J, Tosk JM, Sverd J, Gysin R, Flanagan SD (1991) The dopamine D2 receptor locus as a modifying gene in neuropsychiatric isorders. Journal of the American Medical Association 266:13:1793-1800
Courchesne E, Hesselink JR, Jernigan TL, Yeung-Courchesne R (1987) Abnormal neuroanatomy in a nonretarded person with autism. Unusual Findings with Magnetic Resonance Imaging. Archives of Neurology 44:3:335-341
Courchesne E, Kilman B, Galambos R, Lincoln A (1984) Autism: processing of novel auditory information assessed by event-related brain potentials. Electroencephalography and Clinical Neurophysiology 59:3:238-248
Courchesne E, Lincoln A, Kilman B, Galambos R (1985) Event-related brain potential correlates of the processing of novel visual and auditory information in autism. Journal of Autism and Developmental Disorders 15:1:55-76
Courchesne E, Lincoln A, Yeung-Courchesne R, Elmasian R, Grillon C (1989) Pathophysiologic findings in nonretarded autism and receptive developmental language disorder. Journal of Autism and Developmental Disorders 19:1:1-17
Courchesne E, Press G, Yeung-Courchesne R (1993) Parietal lobe abnormalities detected with mr in patients with infantile autism. American Journal of Roentgenology 160:2:387-393
Courchesne E, Saitoh O, Yeung-Courchesne R, Press G, Lincoln A, Haas R, Schreibman L (1994) Abnormality of cerebellar vermian lobules VI and VII in patients with infantile autism: Identification of hypoplastic and hyperplastic subgroups with MR imaging. American Journal of Roentgenology 162:1:123-130
Courchesne E, Townsend J, Saitoh O (1994) The brain in infantile autism: Posterior fossa structures are abnormal. Neurology 44:2:214-223
Courchesne E, Yeung-Courchesne R, Press G, Hesselink JR, Jernigan TL (1988) Hypoplasia of cerebellar vermal lobules VI and VII in autism, New England Journal of Medicine 318:21:1349-1354
Crepel F (1982) Regression of functional synapses in the immature mammalian cerebellum. Trends in Neurosciences 5:266-269
Crispino L, Bullock TH (1984) Cerebellum mediates modality-specific modulation of sensory responses of the midbrain and forebrain in rat. Proceedings of the National Academy of Sciences 81:9:2917-2920
Damasio A, Maurer R (1978) A neurological model for childhood autism. Archives of Neurology 35:12:777-786
Daniels WW, Warren RP, Odell JD, Maciulis A, Burger RA, Warren WL, Torres AR (1995) Increased frequency of the extended or ancestral haplotype B44-SC30-DR4 in autism. Neuropsychobiology 32:3:120-123
Davila-Garcia MI, Azmitia EC (1989) Effects of acute and chronic administration of leu-enkephalin on cultured serotonergic neurons: Evidence for opioids as inhibitory neuronal growth factors. Developmental Brain Research 49:1:97-103
DeLong GR, Bean SC, Brown FR (1981) Acquired reversible autistic syndrome in acute encephalopathic illness in children. Archives of Neurology 38:3:191-194
DeLong GR, Nohria C (1994) Psychiatric family history and neurological disease in autistic spectrum disorders. Developmental Medicine and Child Neurology 36:5:441-448
DeLong GR (1992) Autism, amnesia, hippocampus, and learning. Neuroscience and Biobehavioral Reviews 16:1:63-70
Deonna T (1991) Acquired epileptiform aphasia in children (Landau-Kleffner Syndrome). Journal of Clinical Neurophysiology 8::288-298
Egaas B, Courchesne E, Saitoh O (1995) Reduced size of corpus callosum in autism. Archives of Neurology 52:8:794-801
Filipek PA (1995) Quantitative magnetic resonance imaging in autism: The cerebellar vermis. Current Opinion in Neurology 8:134-138
Filipek PA, Richelme C, Kennedy DN, Rademacher J, Pitcher DA, Zidel S, Caviness VS (1992) Morphometric analysis of the brain in developmental language disorders and autism (abstract). Annals of Neurology, 32:475
Folstein S, Rutter M (1977) Infantile autism: A genetic study of 21 twin pairs. Journal of Child Psychology and Psychiatry and Allied Disciplines 18:4:297-321
Gaffney GR, Tsai LY, Kuperman S, Minchin S (1987) Cerebellar structure in autism. American Journal of Diseases of Children 141:12:1330-1332
Gao J-H, Parsons LM, Bower JM, Xiong J, Li J, Fox PT (1996) Cerebellum implicated in sensory acquisition and discrimination rather than motor control. Science 272:545-547
Garber HJ, Ritvo ER (1992) Magnetic resonance imaging of the posterior fossa in autistic adults. American Journal Psychiatry 149: 245-247
George MS, Costa DC, Kouris K, Ring HA, Ell PJ (1992) Cerebral blood flow abnormalities in adults with infantile autism. Journal of Nervous and Mental Disease 180:7:413-417
Gillberg C (1986) Onset at age 14 of a typical autistic syndrome. A case report of a girl with herpes simplex encephalitis. Journal of Autism and Developmental Disorders 16:3:369-375
Gillberg C, Trygstad O, Foss I (1982) Childhood psychosis and urinary excretion of peptides and protein-associated peptide complexes. Journal of Autism and Developmental Disorders 12:3:229-241
Gillberg C, Terenius L, Lonnerholm G (1985) Endorphin activity in childhood psychosis: Spinal fluid in 29 cases. Archives of General Psychiatry 42:8:780-783
Gillberg IC (1991) Autistic syndrome with onset at age 31 years: Herpes encephalitis as a possible model for childhood autism. Developmental Medicine and Child Neurology 33:10:920-924
Haas RH, Townsend J, Courchesne E, Lincoln AJ, Schreibman L, Yeung-Courchesne R (1996) Neurologic abnormalities in infantile autism. Journal of Child Neurology 11:84-92
Hallett M, Lebiedowska MK, Thomas SL, Stanhope SJ, Denckla MB, Rumsey J (1993) Locomotion of autistic adults. Archives of Neurology 50:12:1304-1308
Harter MR, Anllo-Vento L (1991) Visual-spatial attention: Preparation and selection in children and adults. In: Brunia CHM, Mulder G, Verbaten MN (eds) Event-related brain research. Elsevier, New York, p 183
Harter MR, Miller SL, Price NJ, LaLonde ME, Keyes AL (1989) Neural processes involved in directing attention. Journal of Cognitive Neuroscience 1:3:223-237
Hashimoto T, Tayama M, Miyazaki M, Murakawa K, Kuroda Y (1993) Brainstem and cerebellar vermis involvement in autistic children. Journal of Child Neurology 8:2:149-153
Hashimoto T, Tayama M, Murakawa K, Yoshimoto T, Miyazaki M, Harada M, Kuroda Y (1995) Development of the brainstem and cerebellum in autistic patients. Journal of Autism and Developmental Disorders 25:1:1-18
Hauser KF, Gurwell JA, Turbek CS (1994) Morphine inhibits purkinje cell survival and dendritic differentiation in organotypic cultures of the mouse cerebellum. Experimental Neurology 130:1:95-105
Heilman KM, Watson RT, Valenstein E (1985) Neglect and related disorders. In Heilman KM, Valenstein E (eds) Clinical Neuropsychology 2nd ed, New York: Oxford University Press
Heinze HJ, Luck SJ, Mangun GR, Hillyard SA (1990) Visual event-related potentials index focused attention within bilateral stimulus arrays. I. Evidence for early selection. Electroencephalography and Clinical Neurophysiology 75:6:511-527
Hetzler BF, Griffin JL (1981) Infantile autism and the temporal lobe of the brain. Journal of Autism and Developmental Disorders 11:3:317-330
Holttum JR, Minshew NJ, Sanders RS, Phillips NE (1992) Magnetic resonance imaging of the posterior fossa in autism. Biological Psychiatry 32: 1091-1101
Hoon AH Jr, Reiss AL (1992) The mesial-temporal lobe and autism: Case report and review. Developmental Medicine and Child Neurology 34:3:252-259
Hughes C, Russell J, Robbins TW (1994) Evidence for executive dysfunction in autism. Neuropsychologia 32:4:477-492
Jones V, Prior M (1985) Motor imitation abilities and neurological signs in autistic children. Journal of Autism and Developmental Disorders 15:1:37-46
Kemper TL, Bauman ML (1993) The contribution of neuropathologic studies to the understanding of autism. Behavioral Neurology 11:1:175-187
Kim S-G, Ugurbil K, Strick PL (1994) Activation of a cerebellar output nucleus during cognitive processing. Science 265:5174:949-951
Kleiman MD, Neff S, Rosman NP (1992) The brain in infantile autism: Are posterior fossa structures abnormal? Neurology 42:4:753-760
Landau WM, Kleffner FR (1957) Syndrome of acquired aphasia with convulsive disorder in children. Neurology 7:523-530
Leiner HC, Leiner AL, Dow RS (1986) Does the cerebellum contribute to mental operations? Behavioral Neuroscience 100:4:443-454
Leiner HC, Leiner AL, Dow RS (1991)The Human Cerebro-Cerebellar System: Its Computing, Cognitive, and Language SkillsBehavioural Brain Research 44:2:114-128
Lovaas OI, Schreibman L, Koegel R, Rehm R (1971) Selective responding by autistic children to multiple sensory input. Journal of Abnormal Psychology 77:3:211-222
Mangun GR, Hillyard SA (1987) The spatial allocation of visual attention as indexed by event-related brain potentials. Human Factors 29:2:195-211
Mangun GR, Hillyard SA (1988) Spatial gradients of visual attention: behavioral and electrophysiological evidence. Electroencephalography and Clinical Neurophysiology 70:5:417-428
Mangun GR, Hillyard SA (1990) Allocation of visual attention to spatial locations: Tradeoff functions for event-related brain potentials and detection performance. Perception and Psychophysics 47:6:532-550
Marchetti B, Scifo R, Batticane N, Scapagnini U (1990) Immunological significance of opioid peptide dysfunction in infantile autism. Brain Dysfunction 3:346-354.
Martineau J, Barthélémy C, Jouvé J, Muh JP, Lelord G (1992) Monoamines (serotonin and catecholamines) and their derivatives in infantile autism: Age-related changes and drug effects. Developmental Medicine and Child Neurology 34:7:593-603
Middleton FA, Strick PL (1994) Anatomical evidence for cerebellar and basal ganglia involvement in higher cognitive function. Science 266:5184:458-461
Mountz JM, Tolbert LC, Lill BW, Katholi CR, Liu HG (1995) Functional deficits in autistic disorder: Characterization by technetium-99m-HMPAO and SPECT. Journal of Nuclear Medicine 36:7:1156-1162
Murakami J, Courchesne E, Press G, Yeung-Courchesne R, Hesselink J (1989) Reduced cerebellar hemisphere size and its relationship to vermal hypoplasia in autism. Archives of Neurology 46:6:689-694
Newman PP, Reza H (1979) Functional relationships between the hippocampus and the cerebellum: An electrophysiological study of the cat. Journal of Physiology 287:405-426
Novick B, Kurtzberg D, Vaughan H Jr (1979) An electrophysiologic indication of defective information storage in childhood autism. Psychiatry Research 1:1:101-108
Novick B, Vaughan H Jr, Kurtzberg D, Simson R (1980) An electrophysiologic indication of auditory processing defects in autism. Psychiatry Research 3:1:107-114
Ornitz EM (1983) The functional neuroanatomy of infantile autism. International Journal of Neuroscience 19:1-4:85-124
Osborne JG, Kindy MS, Hauser KF (1991) Expression of proenkephalin MRNA in developing cerebellar cortex of the rat: Expression levels coincide with maturational gradients in purkinje cells. Developmental Brain Research 63:1-2:63-69
Osborne JG, Kindy MS, Spruce BA, Hauser KF (1993) Ontogeny of proenkephalin mRNA and enkephalin peptide expression in the cerebellar cortex of the rat: Spatial and temporal patterns of expression follow maturational gradients in the external granular layer and in purkinje cells. Developmental Brain Research 76:1:1-12
Ozonoff S, Pennington BF, Rogers SJ (1991) Executive function deficits in high-functioning autistic individuals: Relationship to theory of mind. Journal of Child Psychology and Psychiatry and Allied Disciplines 32:7:1081-1105
Pandya DN, Karol EA, Heilbronn D (1971) The topographical distribution of interhemispheric projections in the corpus callosum of the rhesus monkey. Brain Research 32:31-43
Panksepp J (1979) A neurochemical theory of autism. Trends in Neurosciences 2:174-177
Petersen SE, Fox PT, Posner MI, Mintun M, Raichle ME (1989) Positron emission tomographic studies of the processing of single words. Journal of Cognitive Neuroscience 1:2:153-170
Piven J, Nehme E, Simon J, Barta P, Pearlson G, Folstein SE (1992) Magnetic resonance imaging in autism: Measurement of the cerebellum, pons, and fourth ventricle. Biological Psychiatry 31:491-504
Piven J, Berthier ML, Starkstein SE, Nehme E, Pearlson G, Folstein S (1990) Magnetic resonance imaging evidence for a defect of cerebral cortical development in autism. American Journal of Psychicatry 147:6:734-739
Piven J, Arndt S, Bailey J, Havercamp S, Andreasen NC, Palmer P (1995) An MRI study of brain size in autism. American Journal of Psychicatry 152:8:1145-1149
Piven J, Arndt S (1995) The cerebellum and autism. Neurology 45:398-399
Posner MI (1980) Orienting of attention. Quarterly Journal of Experimental Psychology 32:1:3-25
Posner MI, Walker JA, Friedrich FJ, Rafal RD (1984) Effects of parietal injury on covert orienting of attention. Journal of Neuroscience 4:7:1863-1874
Posner MI, Walker JA, Friedrich FJ, Rafal RD (1987) How do the parietal lobes direct covert attention? Neuropsychologia 25:1A:135-145
Raichle ME, Fiez JA, Videen TO, MacLeod AM, Pardo JV, Fox PT, Petersen SE (1994) Practice-related changes in human brain functional anatomy during nonmotor learning. Cerebral Cortex 4:1:8-26
Raymond G, Bauman M, Kemper T (1989) The hippocampus in autism: Golgi analysis. Annals of Neurology 26:3:483-484
Reichelt KL, Knivsberg A-M, Lind G, Nødland M (1991) Probable etiology and possible treatment of childhood autism. Brain Dysfunction 4:308-319
Reichelt KL, Knivsberg A-M, Nødland M, Lind G (1994) Nature and consequences of hyperpeptiduria and bovine casomorphins found in autistic syndromes. Developmental Brain Dysfunction 7:71-85
Reiss AL, Feinstein C, Rosenbaum KN (1986) Autism and genetic disorders. Schizophrenia Bulletin 12:4:724-738
Ritvo ER, Freeman BJ, Scheibel AB, Duong T, Robinson H, Guthrie D, Ritvo A (1986) Lower purkinje cell counts in the cerebella of four autistic subjects: Initial findings of the UCLA-NSAC autopsy research report. American Journal of Psychiatry 143:7:862-866
Saitoh O, Courchesne E, Egaas B, Lincoln AJ, Schreibman L (1995) Cross-sectional area of the posterior hippocampus in autistic patients with cerebellar and corpus callosum abnormalities. Neurology 45:2:317-324
Schaefer GB, Thompson JN, Bodensteiner JB, McConnell JM, Kimberling WJ, Gay CT, Dutton WD, Hutchings DC, Gray SB (1996) Hypoplasia of the cerebellar vermis in neurogenetic syndromes. Annals of Neurology 39:3:382-385
Sears LL, Finn PR, Steinmetz JE (1994) Abnormal classical eye-blink conditioning in autism. Journal of Autism and Developmental Disorders 24:6:737-751
Shattock P, Lowdon G (1991) Proteins, peptides and autism part 2: Implications for the education and care of people with autism. Brain Dysfunction 4:323-334
Townsend J, Courchesne E (1994) Parietal damage and narrow "spotlight" spatial attention. Journal of Cognitive Neuroscience 6:3:218-230
Townsend J, Courchesne E, Egaas, B (1996a) Slowed orienting of covert visual-spatial attention in autism: Specific deficits associated with cerebellar and parietal abnormality. Development and Psychopathology 8:3:563-584.
Townsend J, Singer-Harris N, Courchesne E (1996b) Visual attention abnormalities in autism: Delayed orienting to location. Journal of the International Neuropsychological Society
Vilensky JA, Damasio AR, Maurer RG (1981) Gait disturbances in patients with autistic behavior: A preliminary study. Archives of Neurology 38:10:646-649
Wainwright-Sharp JA, Bryson SE (1993) Visual orienting deficits in high-functioning people with autism. Journal of Autism and Developmental Disorders 23:1:1-13
Williams K, Shattock P, Berney T (1991) Proteins, peptides and autism part 1: Urinary protein patterns in autism as revealed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and silver staining. Brain Dysfunction 4:320-322
Williams RS, Hauser SL, Purpura DP, DeLong GR, Swisher CN (1980) Autism and mental retardation: Neuropathologic studies performed in four retarded persons with autistic behavior. Archives of Neurology 37:12:749-753
Zagon IS, Gibo DM, McLaughlin PJ (1990) Adult and developing human cerebella exhibit different profiles of opioid binding sites. Brain Research 523:1:62-68
Zagon IS, McLaughlin PJ (1986a) Opioid antagonist (naltrexone) modulation of cerebellar maldevelopment: Histological and morphometric studies. The Journal of Neuroscience 6:5:1424-1432
Zagon IS, McLaughlin PJ (1986b) Opioid antagonist-induced modulation of cerebral and hippocampal development: Histological and morphometric studies. Developmental Brain Research 28:2:233-246
Zagon IS, McLaughlin PJ (1987) Endogenous opioid systems regulate cell proliferation in the developing rat brain. Brain Research 412:1:68-72
Zagon IS, Rhodes RE, McLaughlin PJ (1985) Distribution of enkephalin immunoreactivity in germinative cells of developing rat cerebellum. Science 227:4690:1049-1051
Zilbovicius M, Garreau B, Samson Y, Remy P, Barthélémy C, Syrota A, Lelord G (1995) Delayed maturation of the frontal cortex in childhood autism. American Journal of Psychiatry 152:2:248-252.